
Si è svolto a Lisbona, il 22 e 23 Ottobre 2018, il trentaquattresimo convegno internazionale della Histiocyte Society.
Sono stati presentati, per l’Italia, i risultati di due progetti di ricerca che AIRI onlus sta supportando.
Ringraziamo il Dottor Arturo Bonometti e la Dottoressa Maria Luisa Coniglio per aver condiviso con la comunità internazionale il loro importante lavoro e per aver condiviso con noi la loro esperienza formativa.
Quest’anno la Dottoressa Elena Sieni non ha presenziato al congresso perché in maternità, e quindi impegnata su altri fronti, ma non ha trascurato di coordinare e seguire a distanza i colleghi intervenuti.
Come ci hanno riferito i partecipanti, il meeting è stato un momento di grandissimo scambio di informazioni ed esperienze su diagnosi e terapia di tutte le malattie istiocitarie. L’edizione del congresso di quest’anno era incentrata su due grandi temi: da una parte la decodifica della cellula che origina le malattie istiocitarie (con alcune interessantissime letture magistrali da parte di esperti internazionali di immunologia); dall’altra la raccolta dei nuovi dati in merito alle terapie mirate, al loro possibile utilizzo e alle ricadute pratiche del loro eventuale ingresso nella pratica clinica.
Si è parlato molto di linfoistiocitosi emofagocitica e del suo rapporto con le immunodeficienze primitive e dei nuovi scenari di terapia, come anche delle forme neurodegenerative di istiocitosi a cellule di Langerhans e di caratterizzazione molecolare delle forme istiocitarie rare.
I progetti portati dal nostro gruppo di lavoro sono stati molto apprezzati. Grandissimo è stato l’interesse dimostrato per la presentazione della Dott.ssa Maria Luisa Coniglio (AOU MEYER, gruppo di ricerca sulle istiocitosi coordinato dalla Dott.ssa Elena Sieni) in merito agli studi condotti sulla linfoistiocitosi emofagocitica.
La Dottoressa Coniglio è stata candidata al NESBIT PRIZE con un abstract valutato tra i primi tre migliori su un totale di circa 100 presentati. Un grande successo!
Di seguito i dettagli del lavoro.
“HEMOPHAGOCYTIC SYNDROME AND PRIMARY IMMUNODEFICIENCIES: Report from the HLH Italian Registry”
In letteratura sono stati riportati diversi casi di associazione tra immunodeficienze primitive (PID) e HLH.
Revisionando retrospettivamente il nostro registro italiano HLH abbiamo trovato 7 pazienti con immunodeficienza primaria e HLH.
Sulla base dei dati riportati in letteratura e dei nostri dati precedenti, grazie ai nuovi metodi di sequenziamento che ci offrono la possibilità di studiare più pazienti per più geni contemporaneamente, abbiamo deciso di esplorare prospetticamente la presenza di mutazioni dei geni associati a PID in pazienti con HLH.
Il 77% dei pazienti della nostra coorte ha varianti in uno o più geni sia correlati a PID che a FHL e la maggior parte di questi pazienti presentava varianti in più di un gene, suggerendo che l’HLH è probabilmente la manifestazione finale di diversi percorsi di disregolazione infiammatoria. Queste alte percentuali suggeriscono un’associazione non casuale tra HLH e PID e un’aumentata suscettibilità a sviluppare la sindrome. Il 43% dei pazienti presentavano varianti esclusivamente in geni PID. Cinque pazienti erano fortemente suggestivi per PID e in almeno due pazienti è stata ridefinita la diagnosi di ALPS suggerendo così di esaminare i pazienti con HLH per i geni PID.
Il lavoro è stato molto apprezzato, oltre che per il peso scientifico dei dati presentati, anche per la loro analisi sistematica e per l’accuratezza nella presentazione delle correlazioni tra dato genetico e fenotipo clinico. Alla presentazione è seguita una animata discussione, legata alla attualissima e importantissima questione del come interpretare la mole di dati che il biologo molecolare fornisce al clinico, oltre al come inquadrare i casi di linfoistiocitosi emofagocitica con caratteristiche di overlapp con le sindromi da immunodeficit.
Allo stesso modo, anche il poster presentato dal Dottor Arturo Bonometti, assieme al gruppo milanese, sulle forme istiocitarie rare associate a neoplasie mieloidi ha suscitato molto interesse ed ha portato a un paio di lunghi e bellissimi confronti con i referenti mondiali per la classificazione e la diagnosi istopatologica delle istiocitosi in merito alle difficoltà nell’inquadramento diagnostico delle forme istiocitarie rare e della attuale necessità di trovare dei segni clinici, istopatologici o genetici, per capire come meglio passare il testimone al clinico, dopo la diagnosi, soprattutto nei casi con evoluzione acuta.
I nostri ricercatori sono certamente tornati a casa ricchissimi di energie, idee, nuovi contatti e con la consapevolezza di quanto preziosi siano lo scambio ma anche l’ostinatezza nel perseguire l’obiettivo di studiare queste malattie rare così incredibilmente complesse.
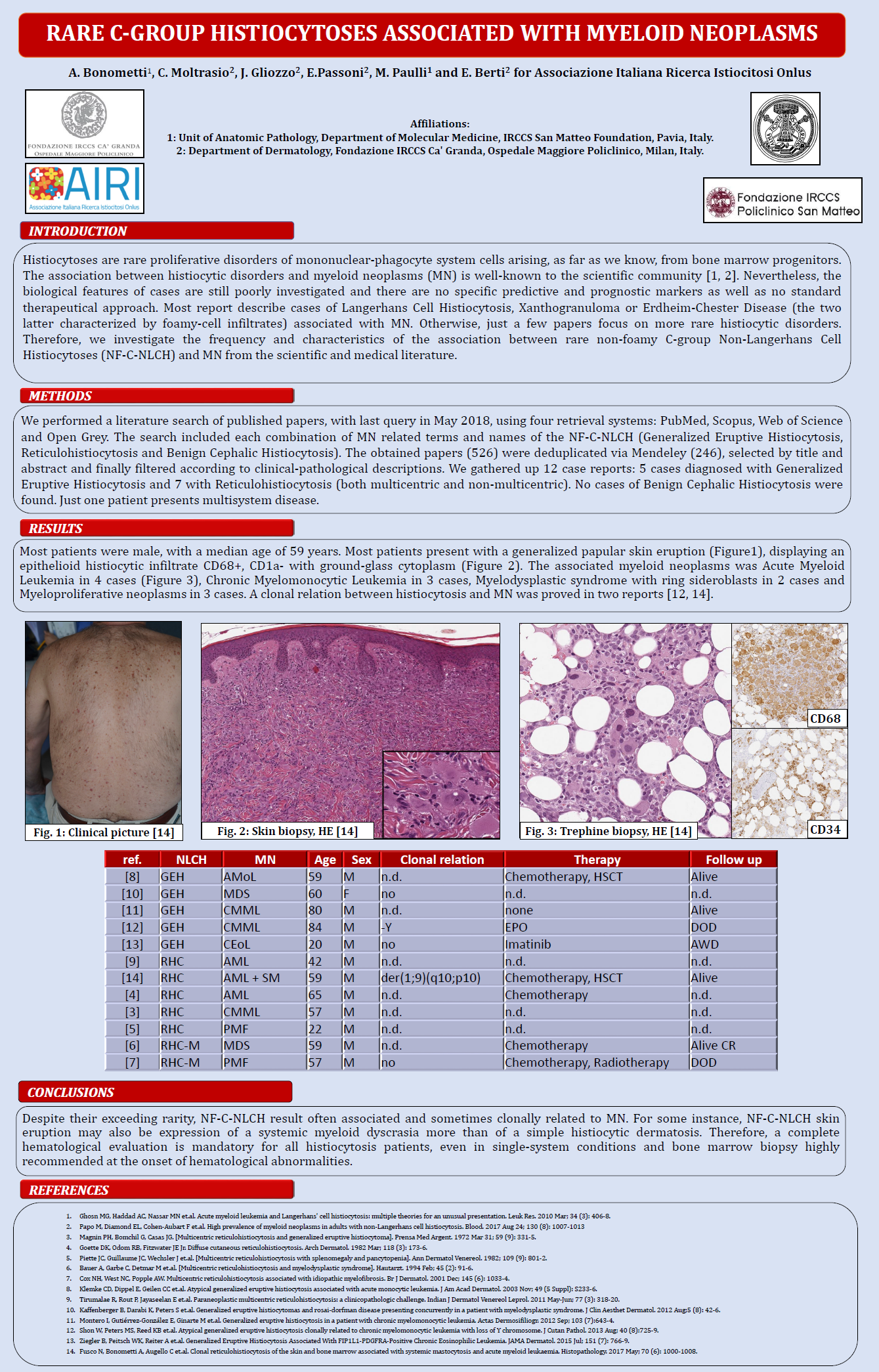
Poster Dottor Bonometti (scarica)


Presentazione Dottoressa Coniglio (scarica)



